吴奇1,2 汪晓辉2 高均2
(1中国科学技术大学化学物理系,科学院选键化学开放实验室,230026)
(2香港中文大学化学系)
引言
1968年,Dusek和Patterson预言[1]:某些聚合物凝胶具有非连续体积相变。近来的研究表明,微小的环境变化诸如温度、pH、电场变化或光照,确能引起一些聚合物凝胶可逆地溶胀与收缩,其体积变化可高达一千倍。这些凝胶具有各种潜在的应用前景,如药物受控释放、选择性吸收、化学记忆,以及制作传感器和人造肌肉等。这些凝胶中一个有趣的例子便是PNIPAM水凝胶[2,3]。在相变点附近,温度变化不到一摄氏度就可引发高达百倍的体积变化。Hirotse等最早用Flory不可压缩晶格模型预言了PNIPAM凝胶的蜷缩(collapse)。可惜这个模型不能完全解释蜷缩温度区内的实验数据,因为它忽略了混合过程中的体积变化以及凝胶网络的拓扑限制。Marchetti等人引入晶格空位及有限链伸展的模型并考虑混合过程中的非零体积变化,从而合理地拟合了PNIPAM凝胶发生较大变形时的数据。Grosberg等人则把凝胶的蜷缩看成是相邻交联点之间的链段(亚链)从伸展到蜷缩(coil-to-globule)的转变,讨论了拓扑限制对这一过程的贡献。这些理论都从某一方面描述了相转变过程。迄今为止,绝大部分实验都基于宏观尺寸的PNIPAM凝胶的溶胀与收缩,并动用了各种实验手段,如显微镜、膨胀计、差示扫描量热计、摩擦测量、小角中子散射及核磁共振分析。然而,有关PNIPAM微凝胶(尺寸小于1μm)的体积相变研究却鲜有报道。Tanaka等人的工作表明,对一颗球形凝胶颗粒,膨胀或收缩所需的时间与颗粒半径的平方成正比。因此,微凝胶对环境变化的响应速度要快得多。不难想象,凝胶的溶胀和收缩是其三维高分子网络中交联点之间的链段的伸展和蜷缩的宏观表现。因此,通过比较水中单根高分子链和微凝胶网络的相转变,作者将能从分子水平上理解凝胶的溶胀和收缩过程。最近,作者对不同形态的PNIPAM,包括单根长链PNIPAM[4~20]、轻度交联的PNIPAM微凝胶[21~25]、表面活性剂与PNIPAM微凝胶的相互作用以及PNIPAM薄膜[26~28],进行了系统研究。这篇综述仅包括了其中与PNIPAM在水中的相变有关的结果。
1 聚(N-异丙基丙烯酰胺)单链在水中的相变
30多年前,利用Flory的平均场理论,Stockmayer[29]首次指出:如果溶剂从良溶剂变成不良溶剂,柔性高分子链可以从伸展的无规线团蜷曲成密度均匀的球。理论和实验都已对这一预言进行了广泛研究[30~32]。由于实验上要求所用大分子的分子量越大越好(通常大于107g/mol),同时,分子量分布(即Mw/Mn)要小于1.1,因此,以往的绝大部分实验都以聚苯乙烯溶液为对象。借助现代的激光光散射等仪器设备和业已健全的光散射理论,实验上已获得许多有用的结果。但是,正如Grosberg和Kuznetsov最近指出的[33],直到1993年,对不含mesogenic基团的不带电的均聚物,还没有一个实验真正观察到热力学稳定的单链蜷缩球体。进而,他们理论上提出了三个问题:(1)能否在单链蜷缩之后且又不互相聚集沉淀的状态下,观察这种单链的蜷缩球体;(2)如果能观察到,那么,在该蜷缩球体中,分子链是否有许多缠结;(3)从单链无规线团到蜷缩球体的动力学过程是怎样的。他们还预言,单链的蜷缩可分成两个动力学过程:链的非缠结快速收缩以及收缩链的缓慢缠结。Chu等人在实验中观察到单根聚苯乙烯链的蜷缩确实可以分成两步。另一方面,从大分子无规线团到蜷缩球体(coil-to-globule)的转变不仅是高分子物理中的一个基本概念,而且还与许多生物的过程有关,如蛋白质分子链的折叠和DNA链的堆积组装。因此,研究水溶性高分子的coil-to-globule转变无疑更有意义。然而,水溶性高分子的coil-to-glob-ule的研究牵涉到更多的相互作用,且大分子量、单分散的水溶性高分子样品的制备极难,见诸报道的研究非常有限。作者注意到聚(N-异丙基丙烯酰胺)(PNIPAM)凝胶在32℃左右有一个十分明显的体积转变,其体积收缩可高达到100倍。此相转变的原动力来自高分子链中憎水和亲水相互作用强弱的变化。这些相互作用比有机溶剂中聚苯乙烯链段之间的vanderWaals相互作用要强得多。因此,水溶液中的PNIPAM链应比有机溶剂中的聚苯乙烯链更容易蜷缩成球状。Kubota[34]等人率先研究了PNIPAM在水中的coil-to-globule转变。证实了最低临界溶液温度(LCST)为32℃,并在PNIPAM链聚集之前观察到高分子单链的有限蜷缩。由于所用PNIPAM样品的分布仍然不够窄(Mw/Mn>1.3),样品中的长链率先蜷缩、聚集,所以,无法真正观察到热力学稳定的蜷缩的球体。Meewes[35]等人则用加入表面活性剂的方法来阻止相分离。可惜,由于表面活性剂的存在,LCST向上移了近3℃,使问题变得更加复杂。为了研究热力学平衡的单链蜷缩球体,作者首先致力制备出窄分布(Mw/Mn<1.05)的高分子量(Mw>1×107g/mol)PNIPAM试样[4,5],并在其基础上,利用光散射系统地研究了极稀溶液(~5×10-6g/mL)中PNIPAM单链的coil-to-globule转变[7~14]。
1.1 理论部分
1.1.1 单链的收缩
对于给定的聚合物溶液,随着温度的改变,溶剂可以从良溶剂变到Θ溶剂,并最终变成不良溶剂。也可以反方向变化。当溶剂性质变差时,柔顺的高聚物链会发生收缩。依修正的Flory理论,链的收缩可以用膨胀因子α≡R(T)/R(Θ)表示,α可从下式[36,37]解出:

1.1.2 Coil-to-globule转变动力学
有关单链紧密球平衡态的理论,很少涉及coil-to-globule转变动力学。这些理论要么是定性描述[33],要么是对有限的链长进行计算机模拟[38]。十年前deGennes指出[39],如果一根聚合物链突然淬冷在不良溶剂中,聚合物的链将采取一种“香肠”状的构型,在链的收缩过程中,每一根“肠”都自行地逐渐缩短并变粗变稠。如果初始动力学是等同的话,用溶剂的粘度数据,可以估计特性驰豫时间为10-3s量级,远小于实验值(102s)。这一差异可以归结为聚合物收缩时产生的局部粘度升高,以及可能存在的拓扑形变限制或紧密球内链的自我缠结。这一切都理所当然地会减缓收缩的动力学过程。基于以上的概念,Grosberg等定性地预言,链的收缩可以分两步走,即一个类似于deGennes描述的快过程和一个类似于自行蛇形行走的慢过程。视觉上可以想象成一个快速皱缩续以缓慢缠结的过程。第一步快过程链的空间密度随链的收缩而迅速增大,第二步伴随着紧密球中链的重排,链密度只是非常缓慢地增加。这两步所对应的驰豫时间τcrum和τeq满足下式:

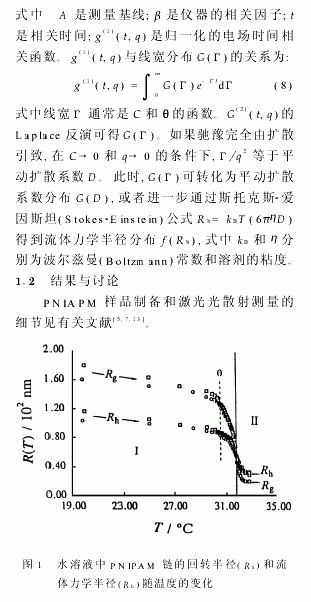
图1显示了PNIPAM链的尺寸的温度依赖性。图中空心圆圈代表样品PNIPAM-1的Rg和Rh;空心方块代表样品PNIPAM-2的Rg和Rh。在区 中,PNIPAM链处于为热力学稳定状态,无聚集。在区 中,PNIPAM链先蜷缩成单链的小球,但这些小球只能在有限的时间(约102min)内稳定存在。随时间推移,它们在热力学趋动下聚集,体系进入两相区。图1表明当温度小于Θ温度时,随T的升高,Rg和Rh线性递减,链轻微地收缩。当温度大于Θ温度时,随着温度逐渐偏离Θ温度,Rg和Rh陡降,链强烈地收缩。由图1可见,当温度从20℃变到33℃时,Rg降低了8倍,Rh降低了3.5倍。Rg与Rh的降幅不同是因为它们本来的定义就不同。Rg与高分子链实际伸展到的空间有关,而Rh仅仅是一个与高分子具有相同的平动扩散系数D的等效硬球的半径。如果高分子链为伸展的高斯(Gauss)链,当高分子链扩散时,链所占空间内的溶剂分子并不全部随链一起运动,所以与高分子链等效的硬球的半径远远小于高分子链实际所伸展到的空间线度。因而Rh比Rg小得多;形成蜷缩的球体后,所含的溶剂分子将随其一同运动,所以等效硬球的半径Rh十分接近蜷缩球体的半径R,而一个均匀球体系的Rg仅为其半径的(3/5)0.5。因此Rg反而小于Rh。

图2显示了当温度从30.59℃分别跃升至31.82℃和33.02℃时,PNIPAM链的蜷缩动力学。升温达到平衡的时间约为400s。当温度从30.59℃跃升到33.02℃时,体系已经处在两相区 中,链的蜷缩过程远快于温度平衡过程,无法跟踪。链的快速蜷缩过程进一步显示链在蜷缩球的内部不可能有高度的缠结,因为链的缠结应当是一个缓慢的过程,尤其是在高度蜷缩的情况下,链的运动已受阻。有趣的是,在温度跃升到31.82℃时(仍在热力学稳定的 区),链的蜷缩(Rh的减小)可分成两步。由于过程太快,无法精确记录,故很难对图2中的数据作定量分析,两步经历的时间分别约为50s和300s。据我们所知,这还是第一次在热力学稳定区域内观察到这样的两步链蜷缩动力学过程。第一步可归结于链的简单收缩,即链的各部分等比例收缩,以至于链在30.59℃时所有的拓扑限制被“冻结”在蜷缩的链中;此时,蜷缩链的构型并没有达到31.82℃时应有的热力学稳定态。因此,第二步反映了蜷缩链的局部调整(“退火”)。图3显示了膨胀因子αs(定义为Rg(T)/Rg(Θ))随相对温度(Θ/T)的变化。当T<Θ时,实验结果与r=10(e5)的Flory理论曲线吻合,环己烷中的聚苯乙烯亦有类似的结果。而当T>Θ时,测定的αs偏离理论曲线。目前,仍然没有一个理论可以解释这一差异。换而言之,在不良溶剂中,理论仍是不完善的。值得注意的是,αs一直下降到一个比方程(1)预计的要低得多的平台值,这可能是因为在水中的憎水和亲水相互作用比理论上预计的要强得多的缘故。在图3中,我们可以看到,αs偏离理论曲线的点可用来精确地测定Θ温度。
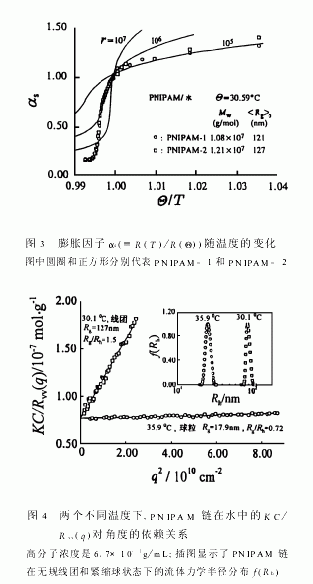
通过进一步完善制样过程,我们得到了在极稀溶液中稳定存在的几乎单分散的高分子量PNIPAM样品,从而第一次观察到热力学稳定的完全蜷缩的单链球。由此我们得以研究和比较其单链在水中蜷曲的相反过程,即从完全蜷曲的单链球到伸展的无规线团的转变。图4分别显示了PNIPAM链的无规线团和完全蜷曲的单链球的KC/Rvv(q)对散射角度的依赖性关系。由方程6知道,图中直线的斜率变化同有关。PNIPAM链的从127nm减小到17.9nm,清楚地反映了链的蜷缩。从无规线团到蜷曲球的尺寸变化也直接反映为图4的插图中所显示的流体力学半径分布曲线(f(Rh))的变化。值得注意的是图4中两条直线在q→0的截距相同,由方程6可知,这说明无规线团和蜷曲球的分子量是一样的,即从无规线团到蜷曲球的相转变是一个单分子过程,确实没有分子间聚集发生。相对应的蜷缩球的f(Rh)分布很窄也说明了这点。由方程6可知:散射光强()∝Mw∝nM2,可知对分子间聚集非常灵敏。实验中,蜷曲球体的溶液的散射光强不随时间变化的事实也说明蜷曲球是稳定的。因此有理由相信,我们第一次从实验上证实了线性均聚物可以从无规线团完全蜷缩为热力学稳定的单链蜷曲球。稍后,我们将进一步阐明在无规线团和单链蜷曲球之间还存在着另外两个热力学稳定状态。

图5和6分别显示了在一个加热-冷却循环中和的温度依赖性。虽然我们已知单个PNIPAM链随着温度升高而迅速收缩的时间一般仅为几百秒[8],图中的每个点仍然都是在样品达到预定温度2h后测得,以确保所测得的和为平衡值。值得提出的是:当达到温度平衡后,即使蜷曲球溶液在对应的温度下放置10h,和仍然没有变化,因为PNIPAM在水中具有最低临界溶液温度(LCST),所以PNIPAM链在加热的过程中会塌缩,而蜷曲链球在冷却过程中会“熔融”。从图5和6中我们发现有两点值得注意:(1)达到蜷曲球的状态区(T>32.3℃),Rg在加热或冷却过程中都不随温度而变化;(2)在转变区(30.5~32.3)℃中,熔融过程中的和均比蜷缩过程中相应的值要小。
在冷却中的滞后现象显示,PNIPAM链从无规线团到蜷曲球的转变是一个不可逆过程。这反映了紧缩球中形成了某些分子内结构,如链内氢键。这些分子内结构的形成阻碍了蜷曲球的“熔融”。只有当温度降到低于25℃时,水成为一个非常好的良溶剂后,这些分子内结构才被完全破坏,和得以回复到在升温前相应的起始值。利用公式<ρ>=Mw/[NA(4/3)π3],我们可以估计链的平均密度(<ρ>)。对应于从无规线团到蜷曲球的转变,<ρ>从0.0025g/cm3。增加到0.34g/cm3,而根据空位填充模型的计算所得的PNIPAM蜷曲球的〈ρ〉约为0.4g/cm3。这意味着每一个PNIPAM蜷曲球所对应的流体力学体积内仍含有66%的水。
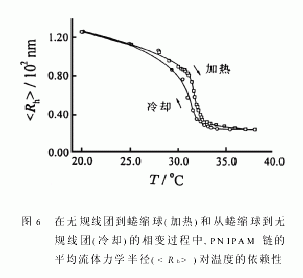
图6还显示在加热过程中,当T>37℃时,已不随温度变化;而在冷却过程中,只有当T<34.0℃时,才开始随温度降低而增加。这意味着在无规线团到蜷曲球的转变中,无规线团逐渐蜷曲成密度均匀的球体;而从蜷曲球到无规线团的转变中,由于塌缩过程中形成的分子内结构使得蜷曲球的融化(swelling)相对较困难。比较图5和6可知,在转变区(30.5℃~32.3℃),比变化得快。这是因为主要依赖于链段密度的径向分布,而相对来说受链内的短程作用影响较大。因为/的比值反映了高分子链的构象特征[41],所以此比值能很好地说明从无规线团到蜷曲球和从蜷曲球到无规线团这两个转变之间的差异。图7显示了在加热过程中,当T<30.6℃时,尽管如图5所示和都随着温度升高而减小,但/不变,反映了链具有良溶剂中无规线团的构象。也就是说,只要在FloryΘ温度以下,高分子链的无规线团构象就保持不变。当30.6℃/急剧地从1.50降到0.56,清楚地反映了链在不良溶剂中的塌缩。在这一转变区域,蜷曲过程可大致分为两个阶段,第一阶段为30.6℃/从1.5降到1.0,反映了从无规线团到“皱缩”线团的构象变化;第二阶段为30.6℃/的实验值比均匀硬球的理论值(3/5)0.5还要小。为了解释这一不寻常的实验现象,让我们仔细考察一下链的蜷缩过程。在一个无规线团中,高分子链会绕成许多杂乱的线圈。当链塌缩时,这些线圈不断变小,从而在蜷曲球的表面形成许多带有应力的小圈,使得球的表面粗糙。从流体力学角度来看,这些小圈中所含的溶液随蜷曲球一起运动,导致增大。但在另一方面,由于组成这些表面小圈的链段数相对很少,故其对的贡献可以忽略。正是由于的增加导致/比均匀硬球的理论预计值(3/5)0.5还要小。这一解释还间接得到了高分子链在均匀胶乳粒子表面吸附的实验所证实。因为,高分子链吸附在粒子表面形成很多圈链(loop),其/值总是小于(3/5)0.5。随着温度升高,链进一步塌缩,最终表面上的小圈缩入球内,形成均匀的蜷缩球。如图5和6所示,减少,但则几乎不变。
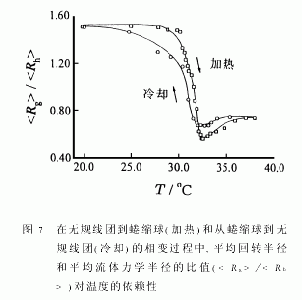
如图7所示,与冷却过程中的转变区域相比,在加热过程的转变区域中,/达到一个更小的值并且融化的蜷曲球存在的温度区间较宽。我们的解释如下:在蜷缩时,应力的增加减慢了表面上小圈的收缩速度,因而导致比减少得慢;而在熔融时,表面的融化要快于因链内结构的形成而受阻的中心,所以比增加得快。综合图5、6和7可见,在/最低点的左侧,/的减小是由于的快速减小,而在最低点的右侧,/的增加则是由于的缓慢减小。最后,值得强调的是,虽然“融化蜷曲球”的概念在蛋白化学中早就存在[42],但对于在主链上没有特殊相互作用的合成均聚物中也存在“融化蜷曲球”,则是作者最近首次在实验上发现的[11]。这一发现说明,“融化蜷曲球”是大分子链的一个普遍存在的物理状态。图7还显示出在熔融冷却过程中,只有当T<25℃时/才接近~1.5。这说明即使水在25℃几乎不变,所以可推论融化球与蜷曲球有几乎相同的链堆积。
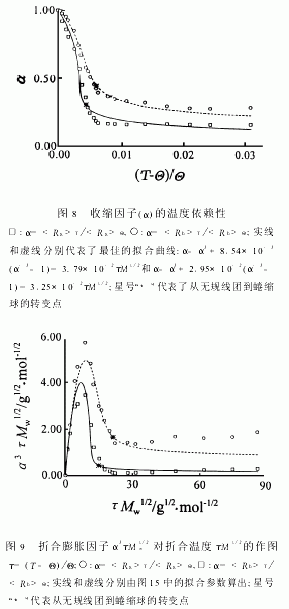
图8显示了收缩因子(α=T/Θ)对增比温度(τ=(T-Θ)/Θ)的依赖性。Birshtein和Pryamistsyn建议α、τ和分子量(M)有以下关系[43]:α-α3+C(α-3-1)=BτM1/2(9)式中B和C是两个当体系给定后不随M和T变化的常数。在方程9中,从无规线团到蜷曲球的自由能的变化由两项组成:(α-α3)对应于高斯链形变的贡献,无扰状态下,α-α3=0;C(α-3-1)对应于分子链内的相互作用,与链所占的体积有关。两项平衡点,即α-α3=C(α-3-1)定义为无规线团到蜷曲球的相转变点,在图中,由星号“*”标出。显而易见,该点比FloryΘ温度高,但却接近停止变化的温度。图中实线和虚线分别代表以B=3.79×10-2和C=8.54×10-3以及以B=3.25×10-2和C=2.95×10-2为参数用方程9拟合的曲线。如图所示,拟合曲线在无规线团区与实验吻合较好 图9反映了α3τM1/2w对τM1/2的依赖。这种依赖关系在聚苯乙烯,聚甲基丙烯酸甲酯和PNIPAM的研究中已有报道[8,32,44]。理论预言,随着T/Θ和T/Θ的减小,α3τM1/2w将分别逼近C/B=2.25×10-1和9.08×10-1。而图9则显示α3τM1/2w在达到最小值后却随着τM1/2w的增加而增加。综合图5、6和9可知,在蜷曲球状态下,和都不随温度而变,所以α3τM1/2w的增加完全是由于τ的增加引致。作者的结果显示,在极稀的溶液中,PNIPAM链在抵达两相区前就已塌缩成完全蜷曲的球并稳定存在。迄今为止的理论都认为在溶液进入两相区前,高分子链会随着溶剂不良性的增加而逐渐收缩。这是因为没有考虑到高分子链具有一定的刚性和直径,不可能象一根理论链那样无限收缩直到相分离发生。实际上,在蜷曲球中,链段的局部密度很高,可能使得单链蜷曲球已进入玻璃态,从而无法进一步收缩。
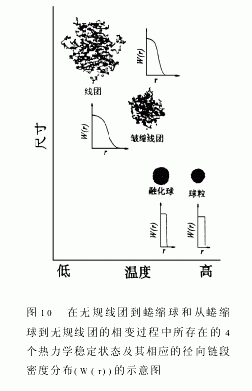
依据我们的实验结果,无论从无规线团到蜷曲球还是从蜷曲球到无规线团的转变都涉及到4个独立的状态:无规线团,皱缩线团,融化蜷曲球和完全蜷曲球。虽然前两个状态可由现存理论来描述,但对融化球蜷曲和完全蜷曲球的定量描述仍然是一个富有挑战性的理论问题。对蜷曲球而言,理论和实验上的偏差,有部分可能来自于图10所示的不同的链段密度分布。定性而言,随着溶剂性质的变坏链段间有效接触的概率会增加;同时,由于链间平均距离的减少,因布朗运动而产生的接触频率也随之增加。链蜷缩过程中,这两个因素的共同作用使得链段间粘结概率逐步增加。即,链蜷缩可能是一个自加速过程。所以,通常实验上观察得到的转变区域要比理论上的计算值窄很多。 |