自交联PM以M水凝胶微球在水介质中的水动力学直径(DH)及其多分散性指数用动态激光光散射(DLLS)(Bl-200 SM型,Brookhaven)测定,激光波长为532 nm,测量角度为900,样品池温度用外置的恒温水浴进行控制,控温精度为士0.01℃,在每一测试温度下的平衡时间为20min,结果取3次测试的平均值。首先测定自交联PNIPAM水凝胶微球在20℃和50℃时的水动力学直径DH,20℃和DH,50℃,然后通过下列公式计算它们的溶胀比a:
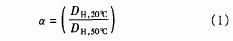
自交联PAIPAM水凝胶微球胶乳样品用真空冷冻干燥机(FDU一2200型,日本理化仪器公司)冻成干粉后,用带固体探头的核磁共振波谱仪(NMR)(Bruker,Av 400)测PNI趾M微球的固体13 C/CwMASNMR谱。样品魔角旋转频率约为5 kHz,使用交叉极化技术,接触时间分别为lms,脉冲间隔时间为4。
2结果与讨论
2.1自交联PNI以M水凝胶微球形成机理的描述
合成自交联PNIPAM水凝胶微球的反应体系中只有单体MPAM、引发剂和水,引发剂一般使用在水中可电离的过硫酸盐引发剂,如APS、KfS(过硫酸钾)等。在60-90℃之间的反应温度下,引发剂分解形成初级自由基,初级自由基很快引发水中溶解的NIPAM单体聚合并进行链增长形成PNIPAM链。PNIPAM链较短时,完全是水溶性的;当PNIPAM链增长到某一临界链长(jcris)时就开始具有温敏性。
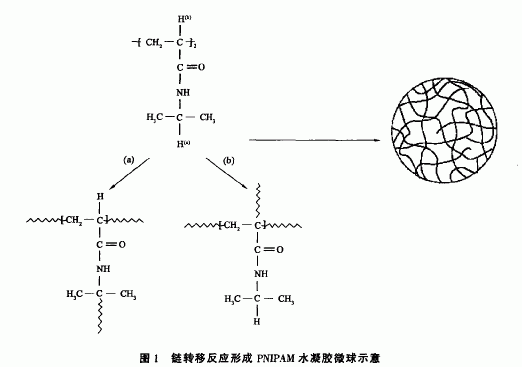
由于反应温度高于PNIPAM的LcST,链长超过jcrit的PMPAM链发生。oil一globule转变从水相中析出,形成基本初始粒子。基本初始粒子粒径很小,表面能很高,极不稳定,很容易发生聚并形成粒径较大的PNIPAM粒子。引发剂分解形成的带电荷的碎片(-SO4)通过共价键连接位于粒子表面,通过静电作用机理为PNIPAM微凝胶粒子提供胶体稳定性。另外由于PNIPAM分子链上含有大量连在叔碳原子上的氢原子,如图1中所示的(a)H和(b)H,反应体系中引发剂分解形成的初级自由基或聚合物自由基均有可能夺取这些H原子,发生链转移反应,使PNIPAM链产生支化结构或形成交联结构(如图1所示)。链长低于jcris的PNIPAM自由基链进人已形成的PNI以M微球内部,或PNIPAM微球内部包埋的自由基都有可能发生同样的链转移反应,使微球内部形成交联网络结构。当体系温度降低到PNIPAM的LcsT以下,形成的微球不会溶解,继续保持胶体粒子状态。
2.2反应温度对自交联州IPAM水凝胶微球粒径、溶胀比和相转变行为的影响
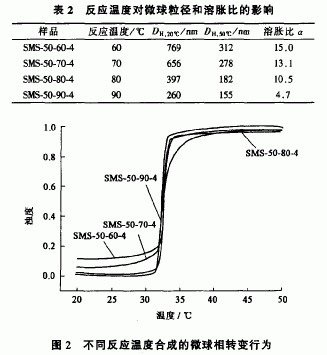
保持单体MPAM用量、引发剂A邢、反应时间以及反应介质pH值不变,改变反应温度合成自交联PNI以M水凝胶微球。它们在温度分别为20℃和50℃时的水动力学直径,以及由此计算的溶胀比大小见表2。随着反应温度升高,自交联PNlpAM水凝胶微球的粒径逐渐减小,溶胀比也明显降低。反应温度越高,引发剂APS的分解速率越快,导致反应体系中自由基浓度增加。反应体系中的自由基除引发单体发生聚合反应使聚合物链增长外,还有可能夺取PM以M聚合物链上的叔碳氢,产生链转移反应,使PMPAM链形成交联结构。反应体系中自由基浓度越高,链转移反应发生的几率越大,PNI-PAM微球中交联密度增加,最终导致微球的溶胀比减小。反应温度越高,反应初期分解的APS摩尔数越多,有更多的一s街离子基团位于初级粒子表面,它们通过静电排斥作用使初级粒子处于比较稳定的胶体状态,而不发生凝聚。这样反应在成核期形成的核数目增多,导致最终PNIPAM微球粒径减小。用紫外一可见光谱仪测得不同反应温度下合成的自交联PNIPAM水凝胶微球水分散体系的浊度随温度变化的关系见图2。从图中可以看出,在任何反应温度下得到的自交联PNIPAM水凝胶微球的相转变温度范围均比过去使用MBA交联剂得到的窄。这可能是由于微球的组成结构中未引人其它组分,同时交联结构也比较均匀。自交联PMPAM水凝胶微球的相转变温度受反应温度影响很小。
2.3反应时间对自交联PNIPAM水凝胶微球粒径、
溶胀比和相转变行为的影响
保持单体NIPAM用量、引发剂A玲、反应温度以及反应介质pH值不变,改变反应时间合成自交联PNI以M水凝胶微球。它们在温度分别为20℃和50℃时的水动力学直径,以及由此计算的溶胀比大小见表3。随着反应时间延长,自交联PNIPAM水凝胶微球的粒径逐渐减小,但趋势不是很明显,而微球溶胀比随反应时间延长而降低的趋势比较显著。这是由于反应时间越长,微球内部形成的交联点越多,导致溶胀比下降。用紫外一可见光谱仪测得不同反应时间下合成的自交联PNIPAM水凝胶微球水分散体系的浊度随温度变化的关系见图3。从图中可以看出,在任何反应时间下得到的自交联PNlpAM水凝胶微球的相转变温度范围都很窄,它们的相转变温度受反应时间影响很小。

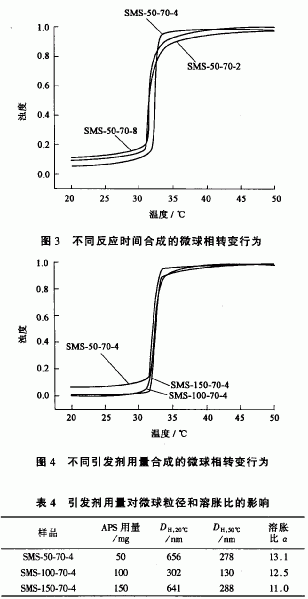
2.4引发剂用量对自交联PNIPAM水凝胶微球粒径、溶胀比和相转变行为的影响
在不用交联剂合成PNIPAM水凝胶微球的情况下,微球的交联网络结构来自于反应体系中自由基发生的链转移反应,因此反应体系中的引发剂用量比使用交联剂的情况下要高得多,否则难以在微球中形成交联网络结构。保持单体NI以M用量、反应时间、反应温度以及反应介质pH值不变,改变引发剂用量合成自交联PM以M水凝胶微球。它们在温度分别为20℃和50℃时的水动力学直径,以及由此计算的溶胀比大小见表4。从表中可以看到,开始随着引发剂用量增加,自交联PMPAM水凝胶微球的粒径减小,但进一步增加引发剂的用量,微球粒径反而增大,可能是由于作为无机盐的APS浓度高,导致反应体系中离子强度增加,尺寸小的微球粒子因静电排斥作用被屏蔽而发生聚集,造成最终PNIPAM水凝胶微球的粒径增大。引发剂用量增加,自交联PNIPAM微球溶胀比的溶胀比减小,但影响没有反应温度那样明显。用紫外一可见光谱仪测得不同引发剂用量的情况下合成的自交联PNIPAM水凝胶微球水分散体系的浊度随温度变化的关系见图4。从图中可以看出,在任何引发剂用量的情况下得到的自交联PNIPAM水凝胶微球的相转变温度范围都很窄,它们的相转变温度也受引发剂用量的影响很小。
2.5自交联PNI以M水凝胶微球结构的表征
自交联PNIPAM水凝胶微球合成过程中没有用到化学交联剂,因此水凝胶微球的形成应该是由于聚合过程中PNI以M链通过链转移反应形成了自交联结构。图1表示出了PNI以M链中最有可能发生链转移反应的两种氢原子:一种是侧链异丙基基团上与叔碳相连的氢原子(a);另一种是主链上与叔碳相连的氢原子(b)。这些氢原子在PMPAM链中相对比较活泼,都有可能被自由基攻击而发生C一H键断裂,形成相对稳定的叔碳自由基。叔碳自由基可能进一步引发MPAM单体发生聚合反应,形成PMPAM支链,也有可能与其它PM以M链上的自由基发生偶合终止反应形成交联点。PNI以M链上形成支化点或PNIPAM链之间形成交联点的结构如图1(a)和(b)所示。为了证实自交联PNI以M水凝胶微球中存在这些支化点和交联点,笔者做了两个样品(样品SMS一50一60一4和sMs一50一70一s)的固体,3cNMR谱。为了进行比较,同时做了线性PM以M聚合物的固体13C NMR谱,结果如图5所示。3个谱图中都出现了3个主要的共振吸收峰,化学位移分别为174 ppm、22 ppm、41 ppm,174 ppm峰和22 ppm峰
分别归属于PNI以M链侧基中的淡基碳原子和甲基原子,41 PPm处的峰是一个比较宽的峰,它应该归属于PNIPAM主链以及侧基中的叔碳原子与仲碳原子,由于固体NMR的分辨率较低,因此很难将它们区分开。124 PPm和223 ppm处的小峰是侧基中拨基碳原子由于实验过程中魔角旋转形成的两个对称的边带峰。比较图5中的3条谱线可以发现,PNIPAM微球[(a)和(b)]的谱线在35 ppm处出现了一个肩峰,而该峰在线性PNIPAM聚合物的谱线中未出现,它应该归属于PNIPAM微球中形成的交联点或支化点上的季碳原子。叔碳原子上的氢原子被另一个碳原子取代后,其化学位移向高场方向移动。比较图5中的谱线(b)和(c)可以看到,谱线(c)中35ppm峰比谱线(b)中的强,可能是由于样品sMs一5。70一8中形成的交联点比样品SMS一50一60一4中的多,这一解释可以通过比较它们的溶胀比来证实。由表2和表3可知,SMS-50-60-4PNIPAM微球的溶胀比为巧.0,而SMS一50一70一8 PNIPAM微球的溶胀比为8 .6,PNI以M微球的溶胀比越小,表明它的交联密度越大,其中交联点越多。至于交联点是按图1中(a)途径形成的多一些,还是按图1中(b)途径形成的多一些,目前的实验结果还不能给出结论,需要进一步对自交联PNIPAM微球的结构进行表征。不过根据与叔碳原子相连的氢原子(a)和(b)所处的化学环境不同,笔者就此还是可以做一些推测:(b)位氢原子被自由基夺取后形成的叔碳自由基因与毅基相连,通过二键的共扼作用而处于比较稳定的状态,因此(b)位氢原子比(a)位氢原子更活泼,所以可推测通过途径(b)形成的交联点应该多一些。
3结论
在没有交联剂存在的情况下,采用无皂乳液聚合法合成出相转变温度范围很窄的自交联PNlpAM水凝胶微球,反应温度、反应时间、引发剂用量对微球粒径和溶胀比有比较明显的影响,这些因素对微球的相转变温度及其范围的影响均很小。固体I3cNMR分析结果表明,PNIPAM水凝胶微球中存在因链转移反应形成的交联点。