陈庆德 沈兴海 高宏成
(北京大学化学与分子工程学院应用化学系 北京 100871)
聚合物微凝胶(简称为微凝胶)为直径在1nm~1μm范围内、具有分子内交联结构的聚合物颗粒[1,2].若在聚合物微凝胶颗粒的表面或内部带有可以进一步反应的基团,即为反应性聚合物微凝胶(简称为反应性微凝胶)[2].Funke等人在此领域进行了系统的、开创性的研究[1~3].由于它具有特殊的流变学性质和在生物医学、涂料与染(颜)料工业、水的净化、石油的采收与输送、胶体或光子晶体的制备、催化剂载体的制备和增加水凝胶强度等领域的优越性和应用前景,微凝胶和反应性微凝胶获得广泛关注[1~11].微凝胶和反应性微凝胶的制备方法有乳液聚合法(包括常规乳液聚合法、无皂乳液聚合法和微乳液聚合法)、分散聚合法和沉淀聚合法,以常规乳液聚合法最为常用[1,2].由于表面活性剂不能与(反应性)微凝胶完全分离,而影响聚合物的性质,使其应用受到限制,故近年来无皂乳液聚合法和溶液聚合法得到广泛关注[4,5].但是,这样得到的(反应性)微凝胶分散液的固含量偏低,在实际应用中有很大的局限性.在Antonietti等[12]重复并优化Staudinger等的实验使聚苯乙烯微凝胶的固含量>10%后,有机溶剂中合成(反应性)微凝胶的固含量问题基本得到解决.然而,在水体系中,Pelton等[13]得到的固含量为2 5%的聚N 异丙基丙烯酰胺微凝胶的结果,仍然没有人能超过.虽然Pitarresi等[14]采用γ 射线辐照技术得到固含量约为5%的无皂聚合物乳液,但其直径的分布为0~35μm,超过了微凝胶直径的上限.辐照聚合法是制备聚合物水凝胶的有效方法之一[15].由于具有不需要任何引发剂、可在室温(甚至玻璃态温度)下进行、加工过程同时起到消毒作用等特点[16],辐照聚合法制备的水凝胶在生物医学方面有着独特的优势.现已有从单体出发、利用辐照法制备聚合物无皂乳液的报道[14,17],但粒径偏大.最近又出现从低浓度的线形聚合物水溶液出发、利用γ 射线辐照[18,19]和脉冲辐解[15,20]引发分子内交联制备微凝胶的报道,但其固含量偏低;若从中等浓度的线形聚合物水溶液出发,则能引发分子间交联而得到宏观水凝胶.N,N′ 亚甲基双丙烯酰胺(Bis)是凝胶和微凝胶合成中较为常用的一种交联剂.4 乙烯基吡啶(4 VP)由于其在制备能吸附甲基橙等有机小分子[21]和汞等重金属离子[22]的聚合物和对pH敏感的微凝胶[23]等领域的特殊性质而逐渐为人所重视.本文详细研究了γ 射线引发4 VP与Bis共聚合成反应性微凝胶的条件,并初步探讨了其机理.
1 实验部分
1.1 试剂
4 乙烯基吡啶(4 VP),含量>96%,Merk,德国,使用前减压蒸馏提纯;N,N′ 亚甲基双丙烯酰胺(Bis)、丙烯酰胺(AM),北京化学试剂公司产品,分析纯,使用前分别用丙酮 苯、氯仿重结晶,常温减压干燥;甲基丙烯酸(MAA)、丙烯酸(AA),分析纯,购自北京化学试剂公司,使用前减压蒸馏提纯.实验用水为3次水.
1.2 聚合反应及凝胶含量的测定
将定量的Bis、4 VP(或MAA、AA、AM)与三次水充分混合后,移入辐照管(直径为15mm)中,控制管中液面高度约为5cm.通高纯N220min以排除体系中的O2后封管,于钴源(剂量率通过硫酸亚铁剂量计测定)中的特定位置照射一定时间.随后,将微凝胶倒入培养皿中,40℃下真空干燥24h后迅速称重(Wb);然后用乙醇、水分别于索氏提取器中抽提24h,40℃下真空干燥24h后迅速称重(Wa).按下式计算凝胶比和溶胶比:凝胶比g=Wa Wb;溶胶比s=1-g
1.3 透射电子显微镜(TEM)观测
将样品原液滴到涂有方华膜的铜网上,自然干燥后用JEOLJEM 200CX型透射电镜观察微凝胶颗粒的形貌.
1.4 动态光散射(DLS)
将微乳胶稀释至适当浓度并经过高速离心(12000r min、30min)后,使用ALV DLS SLS 5022F动态激光光散射仪测量微凝胶的流体力学半径Rh,激光器的波长为632 8nm,测量角度为90°,测量温度为25℃.测量过程中,在保证散射信号可测量的前提条件下,入射光强应尽可能弱.允许的最大误差为±5%.
1.5 热分析
将未经抽提处理的微凝胶充分干燥后,于SetaramDSC131差示扫描量热仪测其DSC图谱.测量过程中通高纯N2吹扫.
1.6 红外光谱
除4 VP采用液膜法外,其余样品均采用KBr压片法,用ThermoNicoletNexusFT IR红外光谱仪测定样品的红外光谱.检测器选用DTGS,样品分析分辨率为4cm-1,扫描次数为64次.
1.7 吸光度
采用ShimadzuUV 2401PC对原液或稀释后的微凝胶在700nm或800nm波长处测定.比色皿采用光程为1cm的玻璃比色皿.
2 结果与讨论
2.1 不同单体对聚合物形态的影响
从表1中可以看出,不同的单体与Bis共聚得到聚合物的性状不同,尤其以4 VP与Bis共聚所得结果最佳,其固含量达到约3.4%时仍然能够得到半透明的乳液,而其它单体与Bis共聚得到的是白色块状凝胶或白色乳液.图1为4 VP与Bis共聚得到的微凝胶的TEM图像,从图中可以看到球形微凝胶颗粒相互重叠,其直径(d)从30到80nm,具有较窄的分布;与DLS的结果(Rh)相比,TEM测得的粒径略小,这可能是微凝胶颗粒在干燥过程中失水收缩的结果.
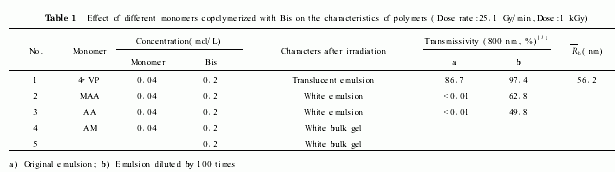

2.2 剂量对微凝胶合成的影响
由于辐照前溶液中Bis的相对含量很大,故只需要约1kGy就可以达到最高凝胶含量(图2a).在凝胶含量较低的区域,s与1 D(D为吸收剂量)具有很好的线性关系(图2a),故可求得s=1时的剂量(即凝胶剂量Dg)为228.8Gy.在凝胶点后,微凝胶的Rh随剂量的增加而减小,然后略有增大(图2b).这可能是凝胶点后、原料完全消耗前所得到的微凝胶具有相对较疏松的结构.随着剂量的增加,富集于微凝胶中未反应的交联剂与剩余双键继续聚合、交联形成一个更加紧密结构,使Rh下降.同时,由于空间因素的制约,在微凝胶颗粒表面有相当数量的双键剩余,在辐照过程中微凝胶颗粒能够通过表面双键聚合而聚.但是由于微凝胶的浓度较小,聚的几率较小,只有在微凝胶颗粒收缩趋于极限后才表现为Rh的略微增大.现已有文献报道微凝胶的转动半径Rg在一定剂量后随剂量增加而下降,而Rh基本保持不变[18];在低浓度下观察到Rg在下降后的轻微上升[20].
由于4 VP和Bis在500~810nm波段无特征吸收峰,此波段的光通过微凝胶的损失来源于微凝胶对光的散射的结果,在测量时表现为微凝胶对光有吸收.由于所得微凝胶颗粒为大质点(2Rh>0 1λ,λ是入射光在介质中的波长),其散射特征与两个参数m、x有关,m为相对折光指数(m=n n0,n和n0是质点及介质的折光指数),x代表质点与波长的相对大小(x=2πr λ,r为质点的半径)[24].由于初始溶液的组成一致,在凝胶点后n变化不大,故可在此波长范围内通过微凝胶的吸光度来定性比较其颗粒的相对大小,即吸光度越大,其粒径越大.实验中选取的入射光波长为700nm.图2b所示,微凝胶的吸光度与Rh有类似的变化趋势(吸收剂量最低的样品的吸光度值较低,这可能是该样品中原料未反应完全,微凝胶浓度较低的缘故).
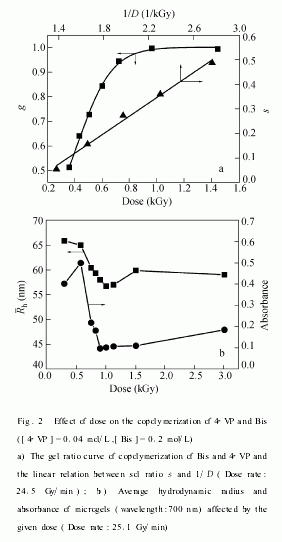
2.3 剂量率对微凝胶合成的影响
在固定总剂量的情况下,改变剂量率,对所得聚合物进行凝胶分析(图3)发现,凝胶含量在较高剂量率时有所下降.这可能是因为,4 VP和Bis混合溶液经辐照后得到的是微凝胶,具有流动性.在较高剂量率条件下,由于自由基的密度较高,易于复合而失去引发聚合的能力,使其能量利用率低于较低剂量率条件下的能量利用率的缘故.从微凝胶的吸光度(图3)可以初步判断,剂量率对微凝胶的尺寸影响不大.
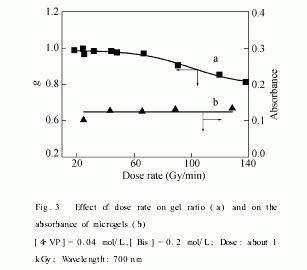
2.4 单体与交联剂比例对微凝胶的影响
从图4中可以发现,无论是保持4 VP和Bis的总浓度一致,增大前者的比例,还是保持Bis的浓度不变,增加4 VP的浓度,微凝胶的Rh均增大.这是由于Bis的相对含量降低时,微凝胶内部交联度下降,其结构相对疏松的缘故.由于初始溶液的组成不一致,在接受相同剂量的辐照后,n变化较大,故不能通过吸光度来定性比较微凝胶颗粒的相对大小.

2.5 聚合物的IR分析
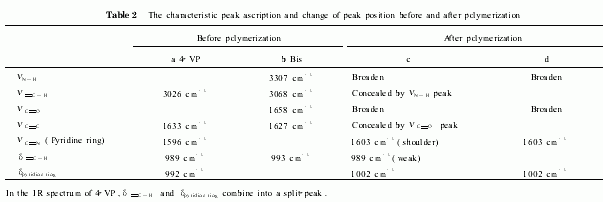
从IR图谱(图5,详细的峰的指认见表2)可以看出,原料的IR图谱明显不同于产物的IR图谱,证实聚合、交联反应确实发生.特别是4 VP在1596cm-1(吡啶环的CN伸缩振动)处的吸收峰在交联聚合后,由于共轭体系变小,而位移至1603cm-1处.Bis在993cm-1(C—H面外摇摆振动)处的吸收峰在交联聚合后变弱,说明聚合后有双键剩余.这是由于空间因素制约,并非所有的双键都参与形成网状结构的缘故,故所得微凝胶为反应性微凝胶.
2 6 聚合物的DSC分析
通过对4 VP与Bis(两者浓度均为0.12mol L)的聚合产物进行DSC分析,没有观察到4 VP线形聚合物的玻璃化转变的信号(线形聚4 VP的玻璃化温度为415K[25]).结合IR的分析结果,可以初步判断为4 VP与Bis在水环境中经辐照共聚得到反应性微凝胶.

2.7 微凝胶形成机理的初探讨
由于4 VP分子中的乙烯基与吡啶环形成共轭体系,聚合时单体自由基或链段自由基上的电子可通过共轭结构转移到吡啶环上,使所得自由基较稳定,使4 VP的聚合反应具有较小的kp和kp/k1/2t值(见表3,kp、kt分别为链增长速率常数和链终止速率常数,聚合速率与kp/ k1/2t成正比),即不易链增长,易导致链终止.另外,Bis的第一双键参与聚合后,它的第二双键的反应活性下降[26].这样,在聚合初期得到支化度较低的短链聚合物.随着4 VP和Bis浓度的降低,短链聚合物开始发生支化,并通过聚合物链中Bis单元的第二双键发生分子内交联反应,在水中的溶解度下降而从水相析出,形成基本初始粒子.基本初始粒子一旦形成,便会捕捉水相中的自由基活性链和富集Bis而继续增长成核,微凝胶的浓度逐步增大.随着吸收剂量的增加,富集于微凝胶中未反应的交联剂与剩余双键继续聚合、交联形成一个更加紧密的结构.由于空间因素的制约,在微凝胶颗粒表面有相当数量的双键剩余,在辐照过程中微凝胶颗粒能够通过表面双键聚合而聚.但是由于微凝胶的浓度较小,聚的几率较小.

表3中,比较本文中与Bis共聚的几个单体的kp和kp/ k1/2t值,可以看出AM>AA>MAA>>4 VP.根据上述分析,与Bis共聚初期得到的聚合物链长度顺序为AM>AA>MAA>>4 VP.AM与Bis共聚初期形成的高分子由于轻微水解生成带少量正电荷,聚合物链中正电荷较集中部分能够较好地伸展;而AA、MAA与Bis共聚初期形成的聚合物链由于羧基部分电离生成的—COO-与—CONH+3发生电荷中和而呈蜷曲线团状[28].这样在Bis与AM共聚体系中,随着链增长反应的进行,Bis和AM的浓度逐渐降低,长链聚合物的支化成为可能.在水中,由于聚合物链较长,在发生分子内交联的同时,还能发生分子间交联,使其在水中的溶解性变差,最终发生相分离而从水相析出.最初生成的凝胶继续与水相中的单体、交联剂进行聚合、交联,反应混合物最终形成宏观凝胶.由于在合成过程中出现相分离,并且凝胶的折光指数与水的折光指数差别较大,使所得凝胶不透明.而在AA、MAA与Bis共聚体系,由于高分子链的蜷曲构象不利于分子间交联,而有利于分子内交联形成微凝胶.
综上所述,在剂量率为25 1Gy min、剂量约为1kGy条件下,4 VP(0 04mol L)与Bis(0.2mol L)的稀水溶液经γ 射线辐照无皂乳液聚合得到固含量约为3.4%、平均流体力学半径为56 2nm的反应性微凝胶.在所选取的与Bis共聚的单体中,以4 VP的效果最佳,并且所得微凝胶的大小可以通过吸收剂量、单体相对含量的改变来进行调控,剂量率对其影响不大.4 VP具有较小的kp和kp/ k1/2t值,有利于得到微凝胶.这对提高水溶液中合成其它微凝胶的固含量具有借鉴意义. |